Una aproximación muy útil en el diagnóstico de estas enfermedades, es considerar la franja de edad en que el paciente comienza los síntomas. Una vez situados en ésta, es importante valorar en qué grupo de síntomas o "síndrome" clínico nos encontramos.
En este momento, la investigación sistémica —tanto clínica como mediante exploraciones complementarias dirigidas— de los diferentes órganos más susceptibles de padecer este tipo de enfermedades (sistema nervioso, órganos neurosensoriales, hígado, corazón, riñón) se hace muy necesaria.
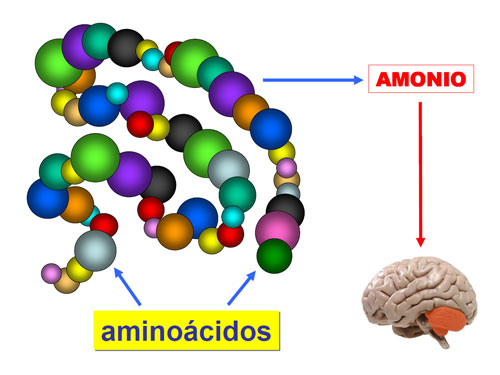
En ocasiones, la presencia de alteraciones bioquímicas características (hiperamoniemia, hipoglucemia, hiperlactacidemia) van a ser imprescindibles para el diagnóstico.
De manera muy sencilla y esquemática podríamos realizar la orientación que detallamos a continuación.
Período neonatal e infancia temprana (<1 año)
De uno a cinco años de vida
Deterioro neurológico progresivo: el enfoque va a depender de cual es el síntoma neurológico predominante y de si hay otros síntomas no relacionados con el sistema nervioso, asociados.
Es frecuente hallar cuadros clínicos en los que predominan:
- Ataxias intermitentes (metabolismo intermediario) o crónicas (mitocondriales, algunas acidurias orgánicas cerebrales, déficit de vitamina E, entre otras),
- Distonías u otros trastornos del movimiento (como en la aciduria glutárica, en el contexto de una regresión con encefalopatía, síndrome de Leigh, o distonías dopa-sensibles en enfermedades de los neurotransmisores, déficits de creatina cerebral),
- Espasticidad (diferentes enfermedades lisosomales como la leucodistrofia metacromática, distrofia neuroaxonal, déficit de arginasa o triple H),
- Epilepsia (como las citadas previamente o algunas enfermedades lisosomales en el caso de epilepsias mioclónicas) o
- Retraso mental asociado o no a trastornos de conducta o del ámbito psiquiátrico (déficit de transporte de creatina, aciduria 4-hidroxi-butírica, algunos trastornos de las purinas, enfermedad de SanFilippo, homocistinurias).
En muchas ocasiones el cuadro clínico es complejo y asocia varios de estos signos.
De cinco años hasta la adolescencia
- Trastorno del movimiento predominante: la enfermedad de Wilson, los déficits de los neurotransmisores o bien los ya mencionados en edades más tempranas.
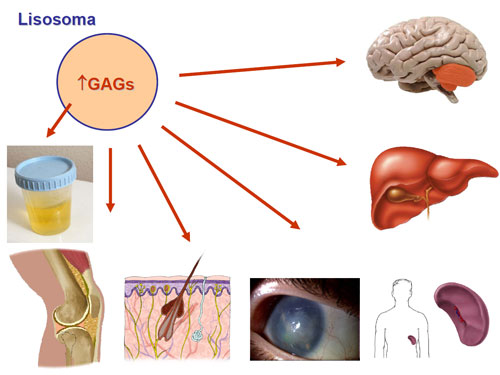
- Deterioro neurológico: en el cual es de utilidad asociar los síntomas previamente descritos en el subgrupo anterior para el diagnóstico diferencial. En esta franja de edad cobran especial importancia las enfermedades lisosomales, la adrenoleucodistrofia ligada al X y formas tardías de defectos del ciclo de la urea.
En cualquier edad
- Ante afectación multiorgánica (+/-dismorfia, +/-displasias óseas) y retraso psicomotor estable o bien que evoluciona hacia un deterioro: pensar en enfermedades por moléculas complejas.
- Existen enfermedades que producen signos clínicos muy específicos que hay que tener en cuenta como el pelo ensortijado (Pili Torti) en la enfermedad de Menkes o acúmulos anómalos de grasa subcutánea en los trastornos de la glicosilación de proteínas, entre otros.
- Hipoglucemias: es importante valorar en qué momento con respecto a la ingesta se producen, si van asociados a cetosis, hiperlactacidemia o hiperamoniemia, así como el tamaño del hígado.
- Hepatopatía, miocardiopatía de causa no conocida.
- La RM cerebral puede ser de gran ayuda: tanto si existe una afectación de sustancia blanca predominante (leucodistrofia), como de sustancia gris (núcleos de la base, atrofia), existen patrones neuroradiológicos de gran ayuda. La RM con espectroscopia informa de la presencia de algunos compuestos químicos del cerebro.