Deficiencia cerebral de creatina. Reunión de la Asociación de Padres con PKU y OTM.

Las deficiencias de creatina cerebral son un grupo de errores congénitos del metabolismo de la creatina de reciente conocimiento.
La creatina es un compuesto natural que tiene un papel importante a nivel energético y de funcionalismo neuronal. El 50% de los requerimientos diarios de creatina para nuestro organismo se obtienen a través de los alimentos (carne, pescados) y el resto a través de una síntesis endógena.
¿Cómo se sintetiza la creatina?
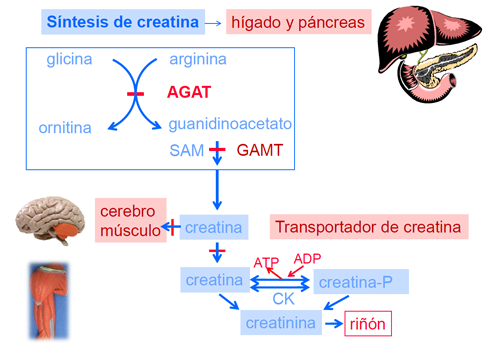
En los seres humanos, la creatina es sintetizada principalmente en hígado, páncreas y riñón, utilizando como sustratos los aminoácidos precursores arginina, glicina y metionina. Son necesarias dos reacciones enzimáticas para formar creatina, la primera de éstas, está mediada por la arginina glicina amidinotransferasa (AGAT) y la segunda por la enzima guanidinoacetato metiltransferasa (GAMT).
El transporte celular es de fundamental importancia en la homeostasis tisular de la creatina. Ésta es transportada hacia los tejidos que posean una elevada actividad de creatinquinasa (CK) -como músculo, cerebro, corazón y testículo-, mediante un sistema de transporte activo (CRTR).
Dentro de la célula, la creatinquinasa interviene en la fosforilación de la creatina formando creatinfosfato, molécula orgánica que en interacción con el ADP forma ATP y actúa a manera de reserva de energía. Finalmente la creatina y creatinfosfato son convertidos mediante una reacción no enzimática en creatinina, la cual se excreta en orina de forma directamente proporcional a la creatina corporal total.
De la fisiología del ciclo de la creatina descrita queda patente su papel en la producción y el almacenamiento energético. Además, dentro de sus principales funciones se encuentra el crecimiento neuronal y se ha demostrado un efecto protector ante la apoptosis celular (muerte celular programada).
Síndrome de deficiencia cerebral de creatina
Los síndromes de deficiencia de creatina cerebral aparecen por alteraciones genéticas que afectan las enzimas participantes en su síntesis (AGAT y GAMT) o a su transportador (CRTR).
En 1994, Stöckler describió la primera enfermedad metabólica relacionada con la creatina: la deficiencia de GAMT. Unos años más tarde (2001) Salomon e Item publicaron los defectos del transportador (CRTR) y la deficiencia de AGAT.
¿Cómo se heredan estas enfermedades?
Las deficiencias enzimáticas de creatina son enfermedades hereditarias, autosómicas recesivas y los genes implicados en ellas se encuentran en el cromosoma 19p13.3 para el GAMT y en el 15q15.3 para el AGAT. El gen para la proteína transportadora se ha denominado SLC6A8 y se localiza en el cromosoma Xq28.
El análisis de estas mutaciones da lugar a la posibilidad de ofrecer consejo genético y realizar diagnóstico prenatal en las familias afectadas.
¿Qué manifestaciones clínicas tienen?
Los pacientes afectos presentan síntomas muy poco específicos y que comparten con un gran número de patologías neurológicas pediátricas.
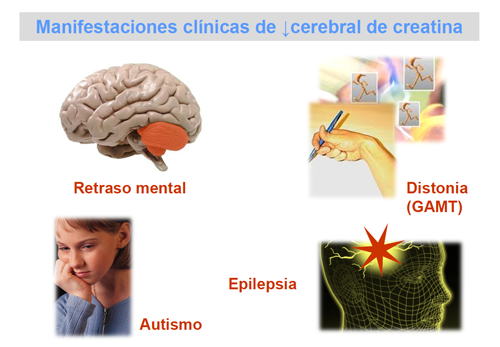
Todos los pacientes con deficiencia cerebral de creatina se caracterizan por un retraso del desarrollo a partir de los 6-12 meses de vida, que se manifiesta posteriormente como retraso mental, problemas de lenguaje y comprensión. El retraso mental y los trastornos del lenguaje son características comunes a los tres defectos, por lo que es muy probable que su causa resida en la deficiencia de creatina cerebral.
Los pacientes con deficiencia de GAMT, con concentraciones elevadas de guanidinoacetato en líquidos biológicos y en tejidos, presentan convulsiones refractarias al tratamiento con antiepilépticos, que complican la enfermedad. Algunos de estos pacientes muestran además trastornos del movimiento e hipotonía muscular.
Las manifestaciones clínicas de los pacientes con defectos del transportador son muy variadas e incluyen: retraso mental de grado variable, epilepsia, autismo y un grave retardo del lenguaje, tanto expresivo como comprensivo.
Diagnóstico
Aún cuando el diagnóstico de los primeros pacientes provenía de la identificación del defecto de creatina a partir de su estudio mediante RMS, este procedimiento es costoso, requiere anestesia (en la mayoría de casos) y además no está disponible en todos los centros.
Por otra parte, la selección de pacientes basada en la clínica debe incluir un amplio espectro de síntomas comunes a muchas otras patologías: retraso mental, trastornos del lenguaje, epilepsia, trastorno generalizado del desarrollo, deterioro neurológico y trastornos del movimiento.
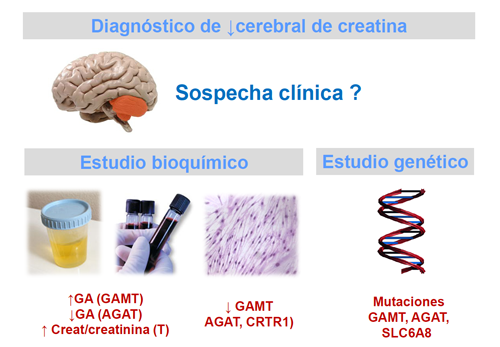
Los pacientes seleccionados con criterios clínicos se estudian bioquímicamente mediante la determinación de guanidinoacetato (GA) y de la relación creatina/creatinina en una muestra de orina de la mañana, mantenida en nevera.
La deficiencia de GAMT se caracteriza por un aumento guanidinoacetato y una disminución de Cr en líquidos biológicos y tejidos. En la deficiencia de AGAT se observa disminución tanto de guanidinoacetato como de Cr. Por el contrario, en la deficiencia de transporte la concentración de guanidinoacetato se halla dentro de los valores control, mientras que la de Cr en orina está aumentada.
Los resultados no siempre son concluyentes y en ocasiones se debe repetir la determinación inicialmente en orina y si confirma de nuevo la alteración en plasma y con la practica de una RM espectroscópica.
La determinación de las actividades enzimáticas de GAMT y AGAT en fibroblastos u otros tejidos confirma los defectos de síntesis, mientras que la incorporación de creatina en fibroblastos permite el estudio del defecto de transporte, tal como hemos podido demostrar en nuestras observaciones.
Tratamiento
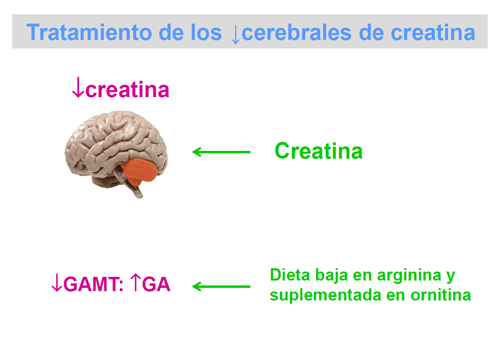
Los pacientes con defectos de síntesis responden positivamente a la administración de monohidrato de creatina. En la deficiencia de GAMT, aún cuando se llega a normalizar la creatina cerebral después de meses de tratamiento, no se logra disminuir la concentración de guanidinoacetato, que es el metabolito al que se atribuye un papel neurotóxico.
La combinación de suplementación de ornitina y reducción de la arginina de la dieta parecen lograr la reducción de la concentración de guanidinoacetato y una cierta mejoría clínica.
Este tratamiento no es válido para los pacientes con deficiencia de transporte, ya que aún después de varios meses de tratamiento, no se detecta creatina o creatina fosfato en cerebro, los síntomas neurológicos persisten, al tiempo que puede constatarse un notable incremento de peso. Ello se debe a que la creatina no atraviesa la barrera hematoencefálica, pero en animales de experimentación se ha demostrado que existe una cierta síntesis cerebral de creatina, lo que hace suponer que estos pacientes podrían responder al tratamiento con arginina.
Estudios terapéuticos en el defecto de transporte de creatina realizados en el HSJD (Dra. Julieta González, HSJD).
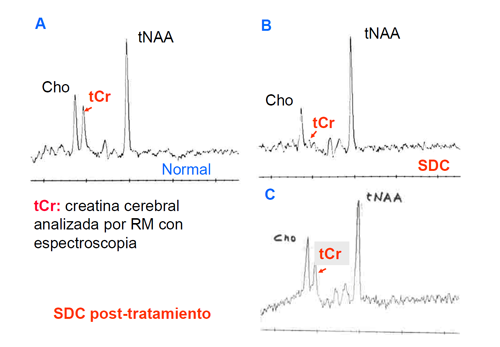
Ante la nula respuesta de estos pacientes al tratamiento con creatina se iniciaron en 2008 una serie de estudios para hallar una terapia eficaz para los defectos de transporte.
En un primer estudio se inició tratamiento con arginina (9 meses) según protocolo de HSJD, con controles clínicos, bioquímicos, neurofisiológicos y neuroradiológicos para evaluar la eficacia de esta medida terapéutica. Aún cuando se producía una elevación de la relación creatina/creatinina y de guanidinoacetato en orina, no se normalizaba la creatina cerebral.
El tratamiento con etiléster de creatina (12 meses) dio lugar a una elevación de la relación creatina/creatinina y de la creatinina en orina, pero no se normalizó la creatina cerebral.
En un siguiente estudio se evaluó el tratamiento con arginina y glicina (9 meses), que no obtuvo resultados clínicos significativos, ni aumento de la creatina cerebral, aun cuando se elevó el índice creatina/creatinina en orina.
Seguidamente se evaluó la dieta cetógena (3 meses), sin cambios significativos.
Estudios terapéuticos en curso (Dras. Antonia Ribes y Angela Arias, IBC-HCB)
Actualmente se trabaja en colaboración con el Institut de Bioquímica Clínica (HCB) + Hospital del Mar (CRBM) para intentar obtener un fármaco que atraviese la barrera hematoencefálica y/o haga las funciones de transportador de la creatina para que alcance el sistema nervioso central.
- 11802 lecturas
También te puede interesar
