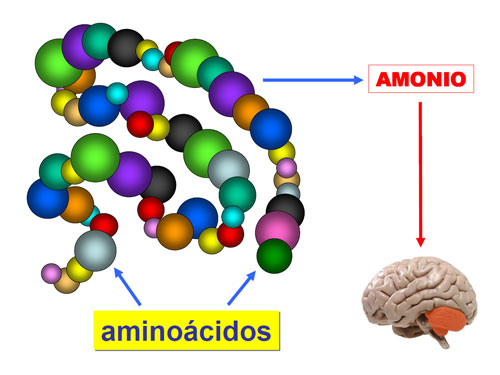
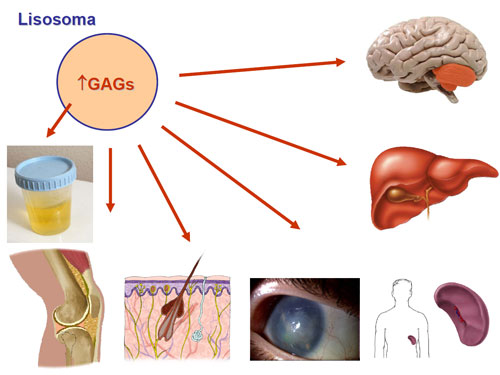
Los principales hallazgos clínicos son: retraso mental, retraso psicomotor, fallo de medro , microcefalia , malformaciones cerebrales, ptosis, cataratas , narinas antevertidas , paladar hendido, cardiopatía congénita, lóbulos pulmonares anómalos, estenosis de píloro, aganglionosis de colon, anomalías renales, anomalías genitales, sindactilia del segundo y tercer dedo del pie, polidactilia. La hipotonía grave de origen central es un signo común, al que contribuye la hipoplasia muscular secundaria a la mala nutrición...